Research
We build statistical and machine learning methods that formalize biochemical principles and expert knowledge into quantitative models. Across our work, the goal is not only prediction, but more interpretable inference: models that make regulatory mechanisms measurable, expose where current assumptions fail, and support better decision-making from noisy or limited genomic data.
Our research is centered on regulatory genomics and transcriptomics, with strong connections to rare disease, common disease genetics, and translational collaborations. A recurring theme is to use mechanistic models of molecular regulation to improve how we connect genomic variation to phenotype.
Rare disease genomics
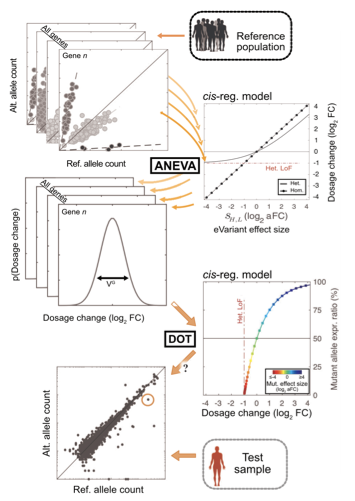
Transcriptome data can reveal pathogenic regulatory effects that are missed by genome or exome sequencing alone. We develop methods that use allelic expression, dosage outliers, and tissue-aware transcriptome analysis to improve molecular diagnosis in rare disease settings, particularly when likely causal coding variants are absent.
These efforts include statistical tests for regulatory aberration, models for prioritizing tissues for transcriptome profiling, and collaborations that keep the work grounded in real diagnostic settings. We collaborate broadly with clinicians and scientists working on pediatric and undiagnosed rare disease cohorts.
Recent work in this area includes diagnostic benchmarking and extensions of the ANEVA framework, including the ANEVA-h preprint.
Selected publications:
- Transcriptomic signatures across human tissues identify functional rare genetic variation.
N Ferraro, B Strober, J Einson, NS Abell, F Aguet, …, P Mohammadi†, S Montgomery†, A Battle†.
Science, 2020. - Genetic regulatory variation in populations informs transcriptome analysis in rare disease.
P Mohammadi†, S Castel, B Cummings, J Einson, C Sousa, …, T Lappalainen†.
Science, 2019. - Improved Identification of Large-effect Rare Genetic Variants using Haplotype Aggregated Allele-specific Expression Data.
K R Ganapathy, M Broly, S Silverstein, M Mendoza, E Song, B Kotis, P Hoffman, PCGC Consortium, A Torkamani, D R Adams, C Bonnemann, T Lappalainen, P Mohammadi.
Preprint, 2025.
Regulatory genomics and effect-size modeling
Most disease-associated loci act through gene regulation rather than protein-coding sequence, but regulatory effects are difficult to interpret with oversimplified models. We develop haplotype-aware and effect-size-oriented models that quantify how cis-regulatory variants change gene dosage, how multiple variants combine, and where current eQTL resources still leave important gaps.
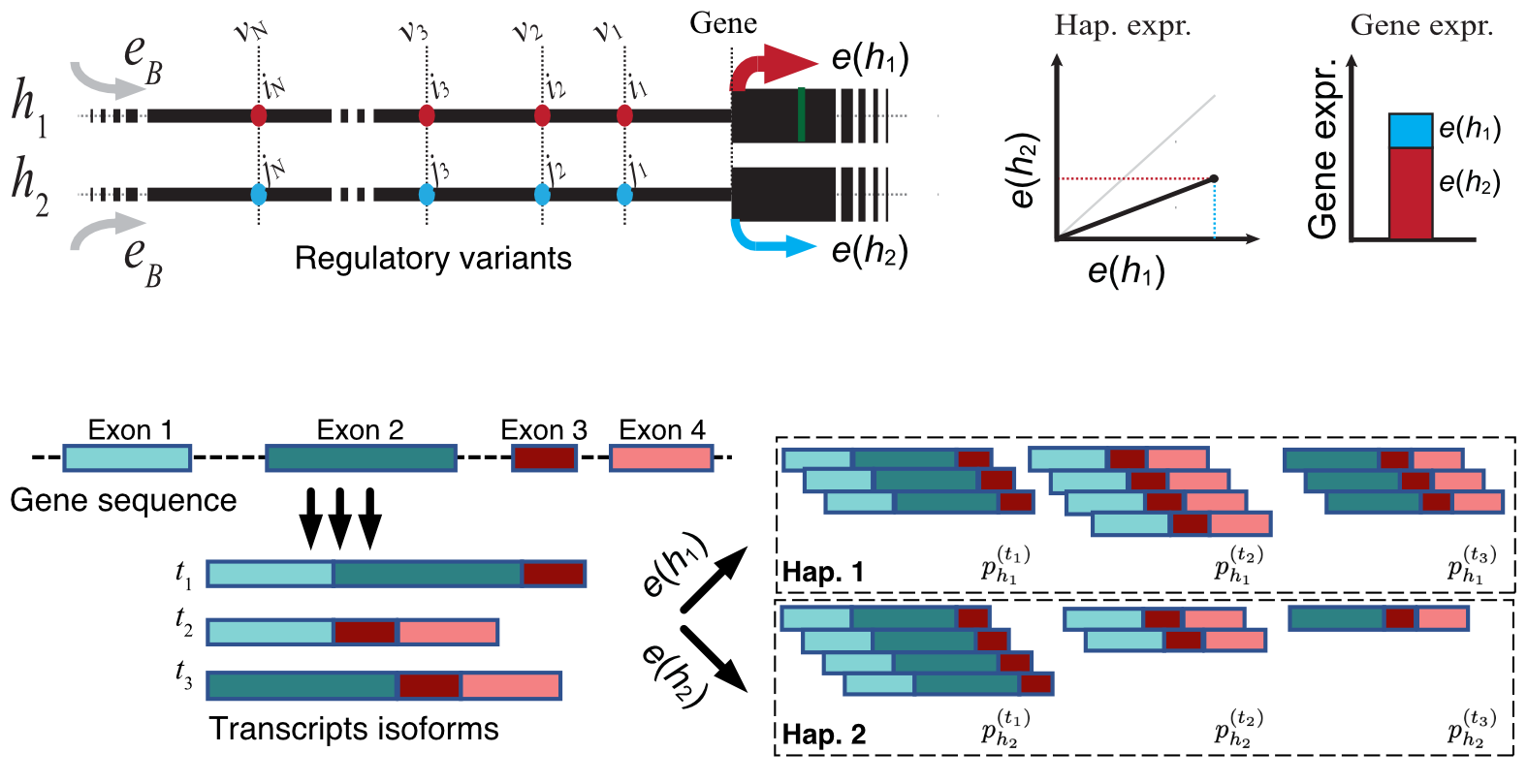
This work draws heavily on large-scale functional genomics resources such as GTEx and related biobank-scale datasets. We use these models both to improve basic understanding of gene regulation and to refine downstream association analyses across diverse populations.
Selected publications:
- Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change.
P Mohammadi, SE Castel, AA Brown, T Lappalainen.
Genome Research, 2017. - Haplotype-aware modeling of cis-regulatory effects highlights the gaps remaining in eQTL data.
N Ehsan, Bence M Kotis, SE Castel, EJ Song, N Mancuso, P Mohammadi.
Nature Communications, 2024.
Multimodal transcriptomics and complex trait genomics
RNA sequencing supports a wide range of phenotypes beyond bulk gene expression, including isoform usage, allele-specific expression, splicing, alternative polyadenylation, transcription initiation, and RNA stability. We develop extensible computational frameworks that turn these measurements into coherent quantitative phenotypes and analyze them jointly with genetic data.
This work is closely tied to our efforts in complex trait genetics. By expanding the set of RNA-derived phenotypes that can be mapped and integrated with association studies, we improve the biological interpretation of GWAS signals and create more informative entry points for TWAS, colocalization, and downstream mechanistic analyses. Recent work in this area includes Pantry and the LaDDR preprint on data-driven RNA phenotyping.
Representative public resources and collaborations include Pantry, RatGTEx, and other large-scale efforts spanning functional genomics, population genetics, and translational genomics.
Selected publication:
- Multimodal analysis of RNA sequencing data powers discovery of complex trait genetics.
D Munro, N Ehsan, SM Esmaeili-Fard, A Gusev, AA Palmer, P Mohammadi.
Nature Communications, 2024. - Data-driven RNA phenotyping captures genetically regulated dimensions of the transcriptome.
Preprint, 2026.
Deep learning, allele-specific signals, and personalized medicine
We are interested in how foundation models and other deep learning systems can be combined with mechanistic and statistical models in integrative genomics, rather than treated as stand-alone predictors. A particular emphasis is allele-specific signal, where personal-genome context, cis-regulatory variation, and transcriptome-derived measurements can be used together to evaluate what these models capture about regulatory function.
This is an active area of work in the lab. We are exploring how sequence-based deep learning and foundation models can be fused with more conventional genomics models to improve interpretation of personal genomes and push the envelope in personalized medicine, while remaining grounded in interpretable biological and statistical structure.