Research
We have a long-standing interest in building statistical methods that systematically incorporate biochemical principles or expert knowledge to effectively extract interpretable results from otherwise limited and noisy experimental data. Our lab applies this strategy to developing quantitative models of genetic regulatory variation. Genetic variation in the regulatory genome plays a major role in human phenotypic variation and disease susceptibility. Currently, our ability to interpret regulatory variation in human genome is hampered by over-simplistic models and limited statistical power. Our efforts to address some of the challenges in interpreting regulatory genome fall broadly into the following two categories:
1. Rare disease diagnostics. Rare diseases are conditions that affect less than 1 in 2000 people in the population. There are >7000 identified rare diseases affecting up to 30 million Americans. The large majority of rare diseases are genetic and manifest early on in life, often with severe health consequences. In more than 60% of the cases in which a rare genetic disease is suspected, whole-genome sequencing fails to identify coding variants that lead to protein truncation or are otherwise potentially pathogenic. Rare variants that affect gene regulation are expected to underlie pathogenesis in some of these cases.
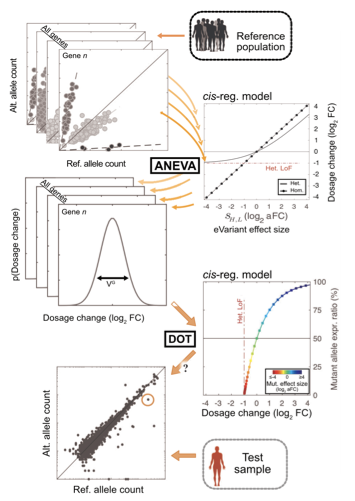
We develop new methods that allow for using transcriptome data to increase diagnostic yield for rare disease patients. These include devising appropriate statistical tests for identifying regulatory aberrant genes and developing mathematical models for identifying the most appropriate tissues for transcriptome profiling in each patient. These efforts are in close collaboration with scientists and clinicians at Neuromuscular and Neurogenetic Disorders of Childhood Section at the NIH, Rady Children’s Institute for Genomic Medicine, and the center for Undiagnosed Disease Network in Stanford university. Below are two of our publications in this area.
- Transcriptomic signatures across human tissues identify functional rare genetic variation.
N Ferraro, B Strober, J Einson, NS Abell, F Aguet, …, P Mohammadi†, S Montgomery†, A Battle†.
Science, 2020. - Genetic regulatory variation in populations informs transcriptome analysis in rare disease.
P Mohammadi†, S Castel, B Cummings, J Einson, C Sousa, …, T Lappalainen†.
Science, 2019.
2. Gene Regulation and Common disease. Around 90% of genomic loci associated with common diseases, such as cancer, type 2 diabetes, or cardiovascular diseases, fall outside the gene boundaries and are thus hard to interpret. In recent years, there has been a deluge of data from genome-wide functional assays. The rapid expansion of computational techniques for mapping genetic correlates of intermediate molecular traits, such as gene expression, has offered opportunities to explore molecular mechanisms that underlie disease susceptibility. Over the past decade, quantitative trait loci mapping studies have identified tens of thousands of common genetic variants affecting the regulation of virtually every protein-coding gene in the human genome. Various biological aspects of these genetic associations, including genomic context and tissue specificity, disease-modifying effects, contribution to common traits, and the effect of local ancestry are established.
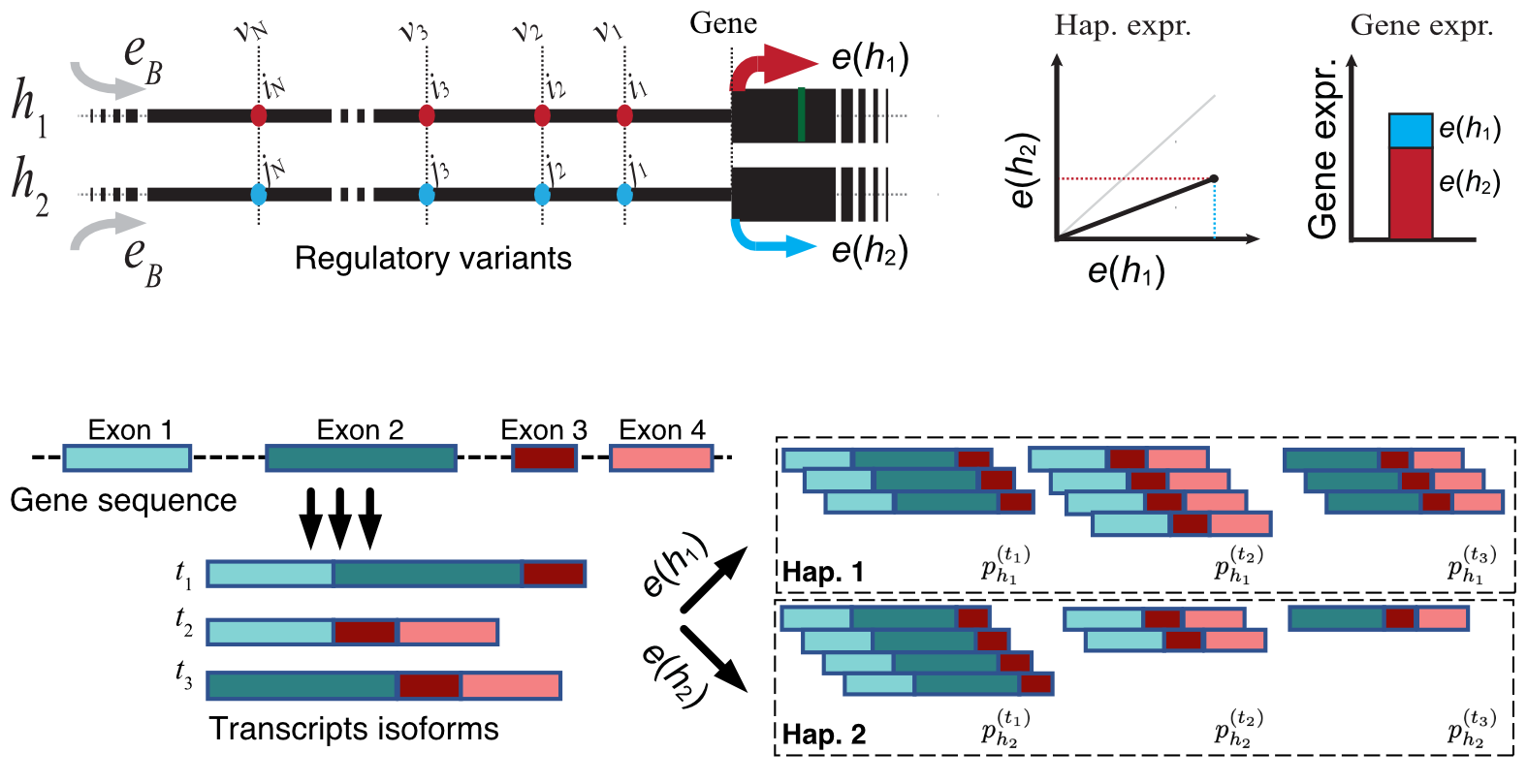
We develop mechanistic models of genetic variation in gene regulation to distill scattered pieces of accumulated knowledge about trends in functional genomic data into unifying theoretical models. These models crystallize our best understanding of the underlying biology and expose the remaining knowledge gaps that remain to be addressed. Using these accurate models of gene regulation, we systematically incorporate the dosage modifying effect of regulatory alleles into genetic association analyses to enhance the resolution of the current genotype-phenotype maps to allow for a more refined mapping of the underlying biological mechanisms that are more generalizable across diverse populations. These efforts are in close collaboration with other scientists in the GTEx consortium and TOPMed project and involve large-scale biobank data from Vanderbilt university bioVU and the UK Biobank. We also contribute to functional analyses of gene expression data (RatGTEx.org) at the national center of excellence for GWAS in Outbred Rats at UC San Diego. Below are a few of our publications in these areas.
- Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change.
P Mohammadi, SE Castel, AA Brown, T Lappalainen.
Genome Research, 2017. - Multimodal analysis of RNA sequencing data powers discovery of complex trait genetics.
D Munro, N Ehsan, SM Esmaeili-Fard, A Gusev, AA Palmer, P Mohammadi.
Nature Communications, 2024. - Haplotype-aware modeling of cis-regulatory effects highlights the gaps remaining in eQTL data.
N Ehsan, Bence M Kotis, SE Castel, EJ Song, N Mancuso, P Mohammadi.
Nature Communications, 2024.